| Митохондриальная миопатия, encephalomyopathy, молочный ацидоз, и инсульт, как эпизоды | |
|---|---|
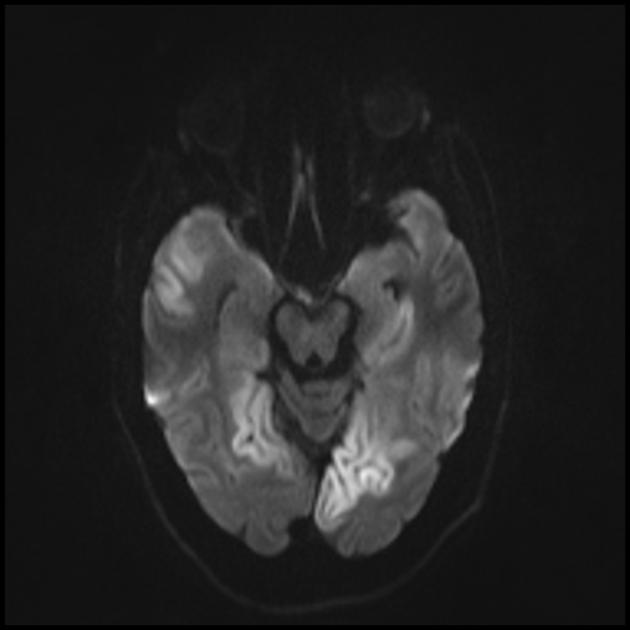
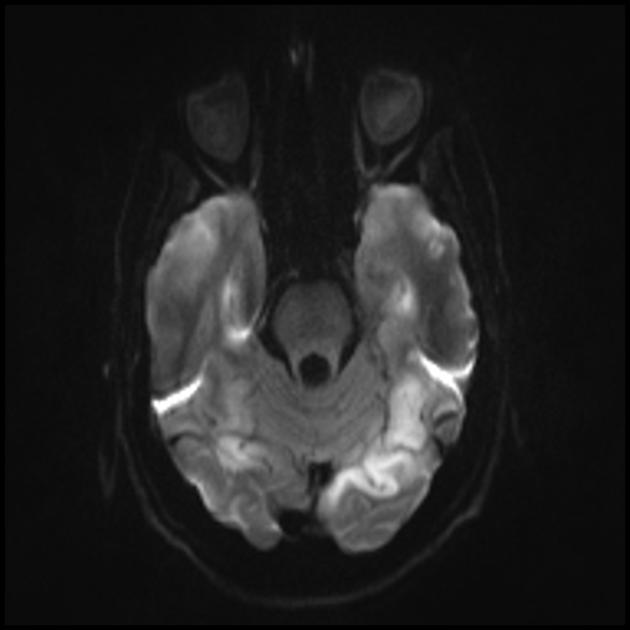
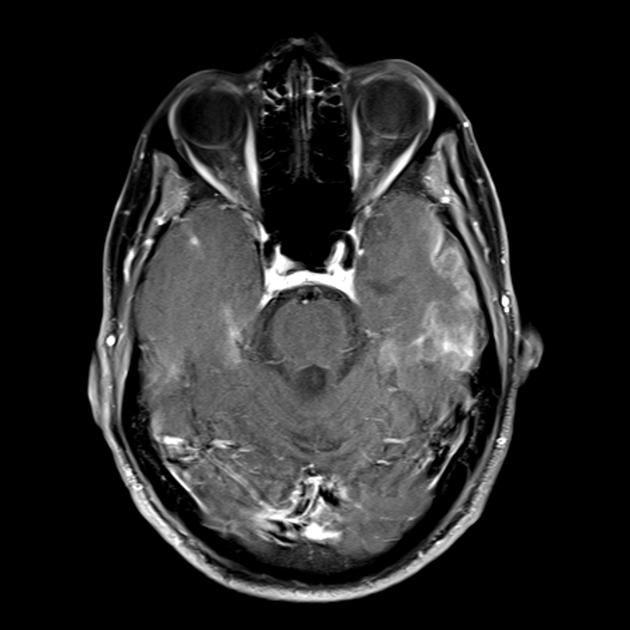
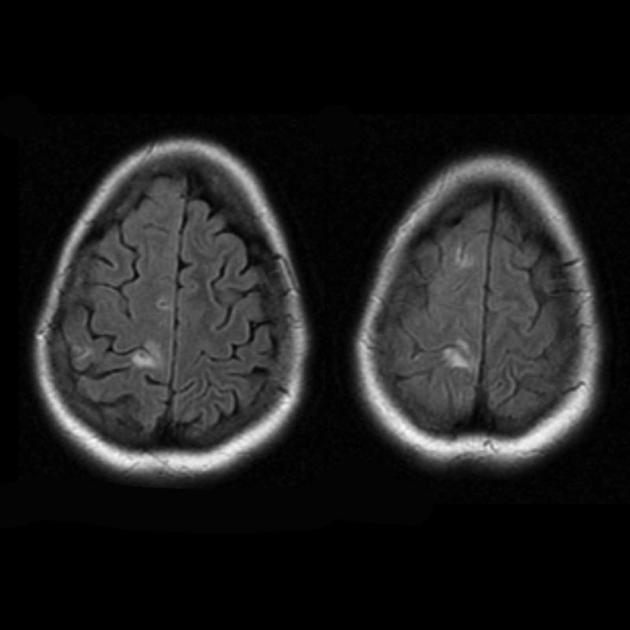
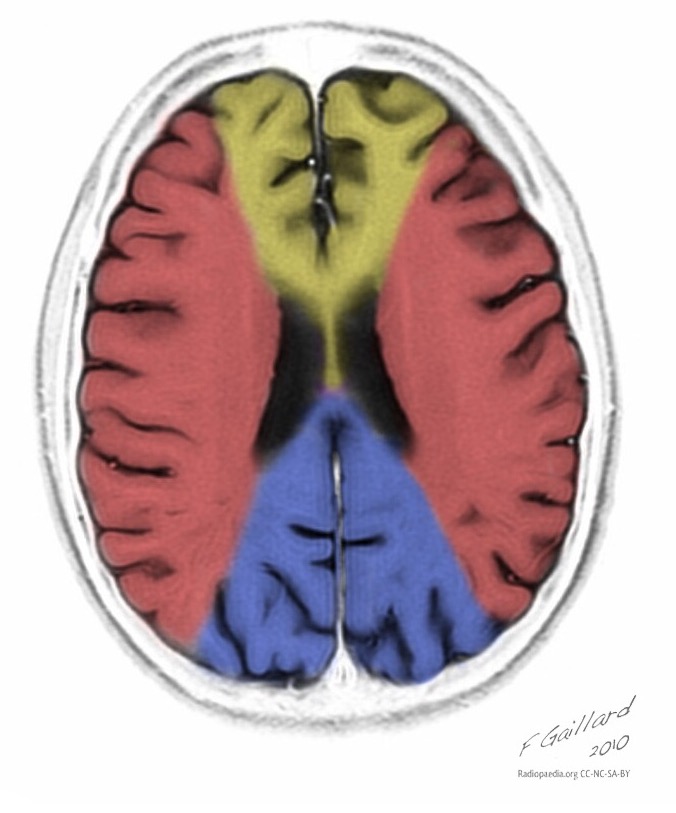
| Базальные ганглии кальцификация, мозжечковая атрофия, увеличение лактата ; КТ изображение человека с диагнозом MELAS | |
| Специальность | неврология |
генетика
Мышечная биопсия человека с диагнозом MELAS , но не несущая известной мутации. (а) Модифицированный Гомори трехцветный краситель показывает несколько рваных красных волокон (стрелки). (б) цитохром с оксидазы пятно, показывающий тип-1 , слегка окрашенных и тип волокна II, темные волокна и несколько волокон с аномальными коллекций митохондрий (стрелки). Обратите внимание, цитохром с оксидазы отрицательные волокна, как правило, рассматривается в митохондриальной энцефалопатии, молочнокислого ацидоза и инсульт-подобные эпизоды (MELAS). (с) Сукцинатдегидрогеназа окрашивание показывает несколько рваных синих волокон и интенсивное окрашивание в митохондриях кровеносных сосудов (стрелка). (г) электронная микроскопия показывает аномальную коллекцию митохондрий с паракристаллическими включениями (стрелок), осмиофильными включениями (большие стрелками) и митохондриальными вакуолями (малые стрелками).
МЕЛАС вызывается мутациями в генах в митохондриальной ДНК.
NADH-дегидрогеназы
Мутации в MT-TL1 причиной более 80 процентов всех случаев MELAS. Они снижают способность митохондрий, чтобы сделать белки, использовать кислород и производить энергию. Исследователи не определили, как изменения в митохондриальной ДНК приводит к специфическим признакам и симптомам MELAS. Они продолжают исследовать эффекты митохондриальных мутаций генов в различных тканях, особенно в головном мозге.
наследование
Это условие наследуются в митохондриальном узоре, который также известен как материнское наследование и гетероплазмии . Эта модель наследования относится к генам, содержащимся в митохондриальной ДНК. Из яйцеклеток, но не сперматозоиды, способствуют митохондрии для развивающегося эмбриона, только самки проходят митохондриальные условия для своих детей. Митохондриальные расстройства могут появиться в каждом поколении семьи и могут повлиять как мужчина, так и женщина, но отцы не проходят митохондриальные черты своих детей. В большинстве случаев, люди с MELAS унаследовать измененный митохондриальный ген от матери. Реже, результаты расстройство от новой мутации в гене митохондриальной и встречается у людей, не имеющих семейной истории MELAS.
диагностика
Лечение / прогноз
Пациенты управляются в соответствии с тем, что участки тела влияют на конкретный момент времени.
Синдром МЕЛАС — это митохондриальное заболевание, характеризующееся поражением мышц и ЦНС.
MELAS (англ. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes - «митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды») - прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии, и сопровождается полиморфной симптоматикой - инсульта, диабетом, судорогами, снижением слуха, сердечными заболеваниями, низким ростом, эндокринопатиями, непереносимостью физических нагрузок и нейропсихиатрическими отклонениями.
История
.
Синдром MELAS был впервые описан в 1984 году Павлакисом и коллегами; десять лет спустя Павлакис и Мицио Хирано опубликовали обзор 110 случаев заболевания.
Тип наследования:
материнский
Эпидемиология :
Точная частота заболевания не известна. В литературе имеются единичные данные о частоте заболевания. На севере Финляндии частота мутации A3243G, составляет 16.3:100 000.
Патогенез :
Мутации митохондральных ДНК, контролирующих дыхательную цепь митохондрий, сопровождаются нарушением процессов окислительного фосфорилирования — важнейшего источника энергии для метаболических процессов в клетке.
Клинические проявления
В возрасте до 40 лет пациенты с МЕЛАС поступают с клиникой транзиторной ишемической атаки, а также с эпилепсией, неоднократной рвоты, головной болью, мышечную слабость. У данных пациентов нередко клинически выявляют деменцию.
Молодой возраст и отсутствие факторов риска, характерных для инсульта, помогает задуматься о МЕЛАС.
Лабораторные данные
Лактат ацидоз — увеличения уровня лактата и пирувата.
Данные визуализации
Изменения головного мозга схожи с изменениями при инсульте.
Отличия от инсульта
1) области поражения не совпадают с границами артериальных сосудистых бассейнов.
2) при повторных приступах очаги визуализируются в другой локализации.
+ клинические данные (молодой возраст, отсутствие факторов риска инсульта).
КТ
Множественные гиподенсивные области не соответствующие сосудистому бассейну.
Кальцификация базальных ганглий (наиболее чаще у пожилых пациентов).
Атрофия возникает на фоне регресса и клинического улучшения.
МРТ
Острый инфаркт
Для дифференциации с инсультом используют ADC и DWI (при инсультах ограничение диффузии (цитотоксический отек), а при МЕЛАС диффузия ограничена незначительно, либо без изменений (вазогенный отек).
Вовлечение в патологический процесс субкортикального белого вещества головного мозга.
Ухудшение визуализации четкости контуров извилин и повышение сигнала от них на Т2-взвешенных изображения.
Хронический инфаркт
Изменения могут быть симметричными и ассиметричными.
Фокальная атрофия возникает на фоне регресса и клинического улучшения.
Теменная, затылочная и височная доля головного мозга наиболее чаще поражаются.
МР-спектроскопия
Повышение уровня лактата.


Синдром МЕЛАС (MELAS) был впервые описан в 1984 году С. Павлакисом и коллегами. Но некоторые исследователи считают, что синдром был уже обозначен ранее такими понятиями, как семейная полиодистрофия, лактацидемия.
Сущность патологии
В 1994 г. С. Павлакис и Мицио Хирано описали 110 случаев заболевания. MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like episodes) - это мультисистемное прогрессирующее нейродегенеративное заболевание. Оно полиморфно и характеризуется энцефалопатией с судорогами и деменцией, лактатацидозом. В основе заболевания лежат мутации митохондриальной ДНК (мтДНК). Данное заболевание имеет и другое название — митохондриальная энцефаломиопатия.
Общие сведения
От 25 до 44 % случаев заболевания носят наследственный характер, передаваясь по материнской линии. В остальных случаях оно возникает впервые. В настоящее время известно более 10 генов, которые мутируют и приводят к развитию данного синдрома. Это гены, кодирующие функции транспортной РНК. Синдром MELAS относится к митохондриальным болезням (МБ) с аномальным скоплением митохондрий, в результате чего нарушается вся система энергетического метаболизма клетки.
Заболевания этой группы передаются только по материнской линии. При них поражаются в разной комбинации наиболее энергозависимые органы и ткани: мышцы скелетные, сердце, мозг, зрение, печень и почки.
Симптоматика полиморфна и может проявляться в любом возрасте. Она включает в себя проявления диабета, сниженного слуха, судорог, эндокринопатий, низкий рост, сердечные патологии, абсолютную неспособность к физическим нагрузкам и психомоторные отклонения.
До нее психомоторное развитие происходит совершенно нормально. Одинаковых по симптомам больных не выявлено, поскольку мутация затрагивает многие гены: MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTTS2, MTND1, 5, 6. Число их продолжает увеличиваться.
У 80 % пациентов синдром MELAS обусловлен точковой заменой A3243G в гене тРНК лейцина (UUR).
Частота изучена недостоверно. Есть только единичные данные: например, в Финляндии частота мутации A3243G составила 16:100 тысячам населения; в Англии - 1 случай на 13 тыс.
Патоморфологические изменения

Характерным патоморфологическим признаком синдрома МЕЛАС являются рваные красные волокна (RRF), которые можно увидеть в мышечной ткани при специальном трехцветном окрасе по Гомори. Они являются следствием мутированных генов и морфологическим субстратом повреждения мтДНК, образуются вследствие пролиферации этих аномальных митохондрий.
Что такое митохондрия вообще
Митохондрии представляют собой двухмембранную органеллу эукариотической клетки (клетки, имеющей ядро), основная функция которой - снабжение энергией. То есть, по сути, митохондрии - это энергетическая база клеток, их энергетические станции.
Количество митохондрий в клетках может меняться на протяжении ее жизни от нескольких штук до тысяч. И больше их бывает в клетках, связанных с выработкой энергии.
Сами митохондрии чаще всего округло-вытянутые, размером от 1 до 10 мкм. Они могут неподвижно застывать или передвигаться внутри цитоплазмы клетки. Перемещаются обычно туда, где требуется больше выработки энергии.
На внутренней мембране митохондрии есть выросты (кристы), на которых имеются целые системы ферментов. В основном, это белковые соединения. Количество крист зависит от интенсивности синтезирующих процессов. Например, в митохондриях мышечных клеток их всегда очень много.
У митохондрий есть автономная система синтеза белка - ДНК, РНК и рибосомы. Часть необходимых белков митохондрии синтезируют сами - 5 %, а часть получают из цитоплазмы - 95 %. Энергия извлекается из органических соединений путем множества ферментативных реакций.
Некоторые эти реакции идут с участием кислорода, т. е. происходит окисление, а после других выделяется СО 2 с переносом протонов водорода и выделением энергии. Если сказать иначе, то митохондрия - это активная участница клеточного дыхания.
Происходят эти реакции на кристах или в самой митохондрии, которая настолько важна для клетки, что если ее вылечить, то клетка полностью будет здоровой.
Патогенез

На первый взгляд при синдроме MELAS состояние напоминает вариант послеинсультного. Но на самом деле он атипичен: возникает у молодых людей, часто провоцируется инфекционными заболеваниями, может протекать в виде злокачественной головной боли, похожей на мигрень, судорог.
Ангиография не дает никаких сосудистых патологий. Могут быть нормальные сосуды или увеличены калибры некоторых артерий, вен, или возникает капиллярная гиперемия.
На МРТ видно, что острые повреждения мозга при синдроме МЕЛАС способны мигрировать и даже исчезать. Некоторые очаги флюктуируют. Для типичного инсульта это совершенно нехарактерно.
При синдроме MELAS имеется наличие мультифокальных некрозов. По большей части это заметно в затылочной части (задняя локализация) коры больших полушарий и белом веществе подкорки. Но могут возникать и в других участках мозга. Участки эти напоминают некроз при инфаркте, но находятся вне бассейнов центральных мозговых сосудов.
Симптоматические проявления

Обычно синдром МЕЛАС у детей дебютирует в возрасте 6-10 лет (может начаться и в 3 года, и в 40 лет). Раннее начало заболевания более типично и коснулось 90 % пациентов. При раннем возникновении заболевание течет более сложно. Больные обычно низкорослые, мышечно слабые и абсолютно не приспособлены к физическим нагрузкам.
Любое напряжение или двигательная активность ухудшает самочувствие. Из внутренних органов поражается сердце с нарушениями питания мышцы и проводимости с последующим развитием сердечно-сосудистой недостаточности. Также возникает нефропатия, диабет, нарушения ЖКТ с рвотой, слух снижается. Характерна мышечная боль, отсутствие рефлексов, парезы, судороги, ИПЭ, потеря сознания. Мышечная слабость (миопатический синдром) и нейросенсорная тугоухость тоже типичны для такой патологии.
Эндокринопатии представлены не только сахарным диабетом, но и недостаточностью гормона роста. Сердечные и почечные нарушения встречаются при развитии рассматриваемого заболевания редко.
Судороги при синдроме MELAS очень вариабельны. Они могут быть фокальными, генерализованными, тонико-клоническими и миоклоническими. Характерна абсолютная нечувствительность судорог к противосудорожной терапии. Нередко бывает, что врачи ставят диагноз эпилепсии и назначают, например, вальпроевую кислоту. После нее самочувствие резко ухудшается и судороги нарастают, потому что она угнетает митохондрии. Деменция хотя и имеет развитие, но манифестным симптомом она становится редко.
Также характерен при заболевании, но он встречается и при многих других патологиях, в связи с чем служить основанием для диагностики он не может. Только при сочетании его с мигренями, судорогами и/или инсультоподобными явленими можно заподозрить начало синдрома МЕЛАС. Даже такая обширная симптоматика не дает правильного диагноза. Прогрессия процесса происходит по-разному.
Признаки

Отличительный клинический признак синдрома MELAS - инсультоподобные эпизоды (ИПЭ), при которых внезапно возникает неврологическая симптоматика. Для ИПЭ характерна несимметричность очагов поражения. Они могут быть множественными.
Избирательность такой локализации дает и определенные очаговые симптомы:
- гемианопсия (корковая слепота);
- гемипарезы;
- сенсорная афазия (непонимание слов);
- акалькулия (нарушения счета);
- аграфия (нарушения правописания);
- атаксия (нарушения координации произвольных движений);
- изменения сознания.
Такие симптомы, похожие на инсульты, нередко возвращаются каждые 1-3 месяца. Особенность острых эпизодов при МЕЛАСе в том, что они имеют быстрый регресс, но при этом часто рецидивируют, то есть как будто бы проходят без следа. Кроме того, у пациентов с таким заболеванием в базальных ганглиях откладываются кальцификаты (это обнаруживается на КТ).
Инсультоподобные эпизоды чаще развиваются в возрасте 5-15 лет. Они никогда не становятся итогом тромбоэмболии. Ангиопатия при МЕЛАСе обусловлена гиперпролиферацией все тех же митохондрий.
ИПЭ в симптомах проявляется рецидивирующими приступами цефалгии, головокружением, парезами, параличами конечностей, черепномозговых нервов. Человек полностью деморализован.
Лактатацидоз при синдроме МЕЛАС

Главным виновником его становится переизбыток в крови и тканях нервной системы молочной кислоты. При этом резко снижается кислотность крови в артериях. Такой ацидоз - частый спутник сахарного диабета, который имеется при синдроме МЕЛАС.
На ранней стадии проявления неспецифичны. Наблюдаются следующие симптомы: общая слабость, боли за грудиной, апатия, сонливость. Очень характерна миалгия после физической нагрузки и прерывистое учащенное дыхание без всякого запаха.
На средней стадии накапливается молочная кислота и возникает гипервентиляционный синдром (ГВС). В крови скапливается углекислый газ. Начинает формироваться шумное дыхание - Куссмауля. Падает давление вплоть до коллапса, наступает олигурия. Пациент становится беспокойным, бредит, а потом теряет сознание с последующим развитием комы - это уже последняя стадия. Симптомы лактатацидоза развиваются стремительно, а именно, в течение нескольких часов. Затем наступает смерть.
Диагностические мероприятия

Как уже отмечалось, из-за полиморфизма симптомов и мутации большого числа генов диагностика синдрома МЕЛАС затруднена. Проводится:
- общий и биохимический анализы крови;
- биопсия мышц;
- генетическое исследование с проведением сравнительного анализа среди больных родственников;
- КТ головного мозга: зоны инфарктов чаще в гемисферах, реже в мозжечке, базальных ганглиях;
- увеличение калибра сосудов (артерий, вен, капилляров);
- ДНК-диагностика: поиск характерных точечных мутаций в мтДНК.
Методы терапии
Лечение синдрома МЕЛАС не разработано, и он сегодня неизлечим. Есть только попытки замедлить процесс поражения. Лечение идет по двум направлениям: посиндромная терапия (эпилепсии, сахарного диабета) и патогенетическое. Однако эффективной патогенетической терапии сегодня нет.
Существуют симптоматические виды лечения: при ухудшении слуха активно используют слуховые аппараты, при слабости дыхательных мышц оказывается респираторная терапия. Было отмечено, что при синдроме MELAS в крови больного значительно снижается уровень L-аргинина при ИПЭ. Поэтому проводят терапию препаратами аргинина и витаминами. Изучается положительное влияние коэнзима Q или идебинона (нобен), препаратов янтарной кислоты, витаминов К 1 и К 3 , В 2 , В 3 , Е, С; L-карнитина, антиоксидантов (мексидол, милдронат), корректоров лактат-ацидоза (димефосфон) - все они улучшают энергетический метаболизм клетки. При лечении судорог не назначают вальпроаты и барбитураты, поскольку они угнетают митохондрии.
В качестве профилактики этого синдрома лучше всего прибегнуть к методу ЭКО. Если женщина знает, что в роду у нее зафиксирован случай проявления данного заболевания, то цитоплазма для проведения оплодотворения берется от здоровой женщины. Метод пока на стадии изучения, он не массовый.
Некоторые особенности
Диагностирование митохондриальных нарушений требует очень тщательного подхода к терапии. В нее обязательно должны включаться средства метаболического действия. Они стабилизируют процессы тканевого дыхания, окислительного фосфорилирования в клетках. Лишь систематическое проведение подобного лечения может помочь в поддержании состояния больных, предотвращая инсультные эпизоды.
Прогноз
Прогноз неблагоприятен ввиду отсутствия эффективного лечения. Продолжительность жизни с первого появления симптомов обычно не превышает пяти лет. Неизученность причин заболевания ведет к тому, что до сих пор не найдена оптимальная схема лечения. Все это делает шансы на излечение минимальными.
Синдром MELAS (англ. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes — «митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды») — прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии, и сопровождается полиморфной симптоматикой — диабетом, судорогами, снижением слуха, сердечными заболеваниями, низким ростом, эндокринопатиями, непереносимостью физических нагрузок и нейропсихиатрическими отклонениями.
В каждом конкретном случае набор симптомов и их тяжесть может сильно отличаться, поскольку синдром связан с мутациями во многих генах: MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTND1, MTND5, MTND6, MTTS2. Мутации могут возникать впервые у конкретного пациента, либо наследоваться по материнской линии. Всего к 2009 году было обнаружено 23 миссенсных точечных мутаций и 4 делеции мтДНК, приводящих к MELAS, однако продолжаются сообщения о новых пациентах с симптомами расстройства при отсутствии известных мутаций.
Распространённость сложно оценить из-за разнообразия проявлений и связанной с этим трудностью диагностики. Среди взрослого населения Финляндии число лиц с синдромом и мутацией A3243G было оценено в 10.2 человека на 100000. На севере Англии распространённость этой же мутации оценена в 1 на 13000 человек. Как предполагается, митохондриальные заболевания связаны с большой пропорцией нейрогенетических повреждений у взрослых, возможно, являясь одной из наиболее частых причин таких расстройств.
Распространённость собственно мутации A3243G, ответственной за большую часть случаев синдрома, оценивается гораздо выше, но мутация не всегда ведёт к заболеванию, поскольку из-за гетероплазмии большинство митохондрий у человека могут быть "здоровыми".
Тип наследования болезни материнский. У всех больных с синдромом NARP обнаруживается гетероплазмическая мутация T=>G в положении 8993 (ген АТФазы 6 - субъединицы V комплекса дыхательной цепи). Уровень гетероплазмии является решающим для характера манифестации данной мутации: при содержании мутантной мтДНК <78% заболевание может проявляться изолированными расстройствами зрения, несколько более высокий уровень мутантной мтДНК сопровождается развитием различных вариантов синдрома NARP, тогда как у больных с уровнем мутантной мтДНК 90% и более наблюдается драматически иной фенотип с быстрым фатальным исходом - болезнь Лея.
Возраст, в котором манифестирует заболевание, широко варьирует от младенческого до взрослого, однако чаще всего первые симптомы появляются в периоде от 5 до 15 лет. Начало болезни часто характеризуется инсультоподобными эпизодами, злокачественными мигренями или задержкой психомоторного развития. Инсульты локализуются чаще в височной, теменной или затылочной областях головного мозга, сопровождаются гемипарезом и имеют тенденцию к быстрому восстановлению.
Они обусловлены митохондриальной ангиопатией, характеризующейся избыточной пролиферацией митохондрий в стенках артериол и капилляров сосудов мозга. По мере прогрессирования болезни, на фоне повторных инсультов нарастает неврологическая симптоматика. Присоединяются мышечная слабость, судороги, миоклонии, атаксия и нейросенсорная тугоухость. Иногда развиваются эндокринные расстройства (сахарный диабет, гипофизарный нанизм).
Обследование включает проведение биохимических, морфологических и молекулярно-генетических исследований. Наиболее частая мутация - замена А на G в 3243-м положении. В результате инактивируется транскрипционный терминатор, заключённый внутри гена тРНК. Следовательно, в результате однонуклеотидной замены наступает изменение транскрипционного соотношения рРНК и мРНК и снижение эффективности трансляции. На втором месте по частоте стоит мутация Т на С в 3271-м положении мтДНК, приводящая к развитию синдрома MELAS.
Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку - так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
Синдром MELAS (от англ. Mitochondrial Encephalomyapathy, Lactic Acidosis, and Stroke-like episodes) - митохондриалъная энцефаломиопатия, лактат-ацидоз и инсультоподобиые эпизоды - обычно манифестирует в 5-20 лет. Заболевание проявляется в первую очередь острыми инсультоподобными эпизодами с развитием очаговых изменений в затылочной и теменно-височной областях мозга и появлением соответствующей неврологической симптоматики (парезы, корковые расстройства зрения, судороги, кома, приступы головной боли и рвоты и др.). Появление очагов связывают с преходящей дисфункцией окислительного фосфорилирования в паренхиме мозга, а также структурно-метаболическими нарушениями в стенках артериол и капилляров; характерной особенностью таких «смешанных» по генсзу инфарктов мозга является относительно быстрое восстановление.
При синдроме MELAS могут наблюдаться также миопатические проявления (повышенная утомляемость и непереносимость физических нагрузок), демепция, атаксия, дегенерация сетчатки, нейросенсорная глухота, низкорослость, диабет, кардиомиопатия и целый ряд других мультиорганных проявлений. Характерен значительный уровень лактат-ацидоза в крови и спинномозговой жидкости, при биопсии скелетных мышц нередко выявляется феномен «рваных красных волокон». Синдром MELAS наследуется по материнскому типу, однако исключительная вариабельность клинических проявлений может весьма затруднять оценку семейного анамнеза.
У больных с синдромом MELAS описано, как минимум, 8 точковых мутаций в генах мтДНК, причем 5 из них локализованы в различных участках гена тРНК) . Наиболее частой мутацией является замена A->G в положении 3243 (около 80% больных), а в целом мутации указанного гена лейциновой тРНК обнаруживаются почти в 95% случаев MELAS. В редких случаях у больных MELAS описаны точковые мутации в генах других тРНК и гене СОХ Ш-субъединицы IV комплекса дыхательной цепи. Все мутации обнаруживаются в гетероплазмическом состоянии.
Синдром NARP (от англ. Neuropathy, Ataxia, Retinitis Pigmentosa) - невропатия с атаксией и пигментным ретинитом - характеризуется, в соответствии с названием, развитием прогрессирующей периферической невропатии с мышечной слабостью, мозжечковой атаксии и пигментной дегенерации сетчатки. Как и при других митохондриальных энцефаломиопатиях, клиническая картина может быть весьма вариабельной, с наличием или отсутствием у родственников ряда дополнительных симптомов (задержка психомоторного развития, эпилептические припадки, деменция). Исследования на лактат-ацидоз и другие маркеры митохондриальной дисфункции не всегда информативны.
Тип наследования болезни материнский . У всех больных с синдромом NARP обнаруживается гетероплазмическая мутация T=>G в положении 8993 (ген АТФазы 6 - субъединицы V комплекса дыхательной цепи) . Уровень гетероплазмии является решающим для характера манифестации данной мутации: при содержании мутантной мтДНК <78% заболевание может проявляться изолированными расстройствами зрения, несколько более высокий уровень мутантной мтДНК сопровождается развитием различных вариантов синдрома NARP, тогда как у больных с уровнем мутантной мтДНК 90% и более наблюдается драматически иной фенотип с быстрым фатальным исходом - болезнь Лея .
 Оглавление темы "Митохондриальная патология нервной системы":
Оглавление темы "Митохондриальная патология нервной системы":